|
FFPopSim
2.0
Library for Population Genetics
|
Population class for high-dimensional simulations. More...
#include <ffpopsim_highd.h>
Public Member Functions | |
| haploid_highd (int L=0, int rng_seed=0, int number_of_traits=1, bool all_polymorphic=false) | |
| Default constructor. More... | |
| virtual | ~haploid_highd () |
| Destructor. More... | |
| double | get_mutation_rate () |
| void | set_mutation_rate (double m) |
| bool | is_all_polymorphic () |
| vector< poly_t > | get_polymorphisms () |
| vector< poly_t > | get_fixed_mutations () |
| vector< int > | get_number_of_mutations () |
| int | L () |
| int | get_number_of_loci () |
| int | N () |
| int | get_population_size () |
| int | get_generation () |
| void | set_generation (int g) |
| int | get_number_of_clones () |
| int | get_number_of_traits () |
| double | get_participation_ratio () |
| int | set_allele_frequencies (double *frequencies, unsigned long N) |
| Initialize population in linkage equilibrium. More... | |
| int | set_genotypes (vector< genotype_value_pair_t > gt) |
| Initialize the population with genotype counts. More... | |
| int | set_wildtype (unsigned long N) |
| Initialize a wildtype population (00...0) More... | |
| int | track_locus_genealogy (vector< int > loci) |
| Designates as set of loci to have their genealogy tracked. More... | |
| void | add_genotype (boost::dynamic_bitset<> genotype, int n=1) |
| Add the genotype specified by a bitset to the current population in in n copies. More... | |
| int | add_trait_coefficient (double value, vector< int > loci, int t=0) |
| void | clear_trait (int t=0) |
| void | clear_traits () |
| void | set_random_trait_epistasis (double epistasis_std, int traitnumber=0) |
| int | add_fitness_coefficient (double value, vector< int > loci) |
| void | clear_fitness () |
| void | set_random_epistasis (double epistasis_std) |
| int | evolve (int gen=1) |
| Evolve for some generations under the specified conditions. More... | |
| int | bottleneck (int size_of_bottleneck) |
| Cause a bottleneck in the population size. More... | |
| unsigned int | flip_single_locus (int locus) |
| Flip a spin at a specific locus in random individual. More... | |
| void | calc_stat () |
| Calculate trait and fitness statistics and allele frequences. More... | |
| void | unique_clones () |
| Remove duplicate clones. More... | |
| vector< int > | get_nonempty_clones () |
| Obtain a list of the good clones. More... | |
| int | random_clone () |
| Get a random clone from the population. More... | |
| int | random_clones (unsigned int n_o_individuals, vector< int > *sample) |
| Sample random individuals from the population. More... | |
| string | get_genotype_string (unsigned int i) |
| int | distance_Hamming (unsigned int clone1, unsigned int clone2, vector< unsigned int * > *chunks=NULL, unsigned int every=1) |
| int | distance_Hamming (boost::dynamic_bitset<> gt1, boost::dynamic_bitset<> gt2, vector< unsigned int * > *chunks=NULL, unsigned int every=1) |
| Calculate Hamming distance between two sequences. More... | |
| stat_t | get_diversity_statistics (unsigned int n_sample=1000) |
| Calculate diversity in the current population (Hamming distance between pairs of sequences) More... | |
| stat_t | get_divergence_statistics (unsigned int n_sample=1000) |
| Calculate mean and variance of the divergence from the [00...0] bitset. More... | |
| double | get_allele_frequency (int l) |
| double | get_derived_allele_frequency (int l) |
| double | get_pair_frequency (int locus1, int locus2) |
| Get the joint frequency of two alleles. More... | |
| vector< double > | get_pair_frequencies (vector< vector< int > > *loci) |
| Get the joint frequency of two alleles, for a vector of allele pairs. More... | |
| double | get_chi (int l) |
| double | get_derived_chi (int l) |
| double | get_chi2 (int locus1, int locus2) |
| double | get_LD (int locus1, int locus2) |
| double | get_moment (int locus1, int locus2) |
| void | set_trait_weights (double *weights) |
| double | get_trait_weight (int t) |
| double | get_fitness (int n) |
| int | get_clone_size (int n) |
| double | get_trait (int n, int t=0) |
| vector< coeff_t > | get_trait_epistasis (int t=0) |
| stat_t | get_fitness_statistics () |
| stat_t | get_trait_statistics (int t=0) |
| double | get_trait_covariance (int t1, int t2) |
| double | get_max_fitness () |
| void | update_traits () |
| For each clone, recalculate its traits. More... | |
| void | update_fitness () |
| For each clone, update fitness assuming traits are already up to date. More... | |
| int | get_divergence_histogram (gsl_histogram **hist, unsigned int bins=10, vector< unsigned int * > *chunks=NULL, unsigned int every=1, unsigned int n_sample=1000) |
| Get histogram of divergence from the [00...0] bitset. More... | |
| int | get_diversity_histogram (gsl_histogram **hist, unsigned int bins=10, vector< unsigned int * > *chunks=NULL, unsigned int every=1, unsigned int n_sample=1000) |
| Get histogram of diversity in the population (mutual Hamming distance) More... | |
| int | get_fitness_histogram (gsl_histogram **hist, unsigned int bins=10, unsigned int n_sample=1000) |
| Calculate histogram of fitness from traits. More... | |
| int | print_allele_frequencies (ostream &out) |
| Print all allele frequencies into a stream provided. More... | |
| int | read_ms_sample (istream >s, int skip_locus, int multiplicity) |
| Read the output of Hudson's ms and use it to initialize the genotype distribution. More... | |
| int | read_ms_sample_sparse (istream >s, int skip_locus, int multiplicity, int distance) |
| Read the output of Hudson's ms and use it to initialize the genotype distribution. More... | |
Public Attributes | |
| hypercube_highd * | trait |
| vector< clone_t > | population |
| int | carrying_capacity |
| double | outcrossing_rate |
| double | crossover_rate |
| int | recombination_model |
| bool | circular |
| double | growth_rate |
| multi_locus_genealogy | genealogy |
Protected Member Functions | |
| int | get_random_seed () |
| Get a random seed from /dev/urandom. More... | |
| void | produce_random_sample (int size=1000) |
| Produce and store a random sample of the population for stochastic processes. More... | |
| int | mutate () |
| Mutate random clones at random loci (all loci) More... | |
| int | select_gametes () |
| Generate offspring according to fitness (selection) and segregate some for sexual mating. More... | |
| double | relaxation_value () |
| Get the log of the exp-average fitness plus relaxation term. More... | |
| double | get_logmean_expfitness () |
| Get the population exp-average of fitness, used for keeping the population size fixed. More... | |
| unsigned int | flip_single_locus (unsigned int clonenum, int locus) |
| Flip a spin (locus) in individual. More... | |
| void | shuffle_genotypes () |
| int | new_generation () |
| int | partition_cumulative (vector< unsigned int > &partition_cum) |
| Calculate the cumulative partition of sequences into clones. More... | |
| int | provide_at_least (int n) |
| : allocates memory for a sufficient number of clones More... | |
| void | calc_allele_freqs () |
| calculate and store allele frequencies More... | |
| void | reassortment_pattern () |
| Produce a random reassortement pattern. More... | |
| void | crossover_pattern () |
| Choose a number of crossover points and produce a crossover pattern. More... | |
| int | add_recombinants () |
| Pair and mate sexual gametes. More... | |
| int | recombine (int parent1, int parent2) |
| Recombine two genotypes parent1 and parent2 to produce two new genotypes. More... | |
| int | recombine_crossover (int parent1, int parent2, int ng) |
| void | calc_fitness_stat () |
| Calculate and store fitness population statistics. More... | |
| void | calc_trait_stat () |
| Calculate and store trait population statistics and covariances. More... | |
| void | calc_individual_traits (clone_t &tempgt) |
| Calculate traits of the chosen clone. More... | |
| void | calc_individual_fitness (clone_t &tempgt) |
| Calculate fitness of a particular clone. More... | |
| void | calc_individual_traits (int clonenum) |
| void | calc_individual_fitness (int clonenum) |
| void | check_individual_maximal_fitness (clone_t &tempgt) |
| double | get_trait_difference (clone_t &tempgt1, clone_t &tempgt2, vector< int > &diffpos, int traitnum) |
| Calculate trait difference between two clones. More... | |
| virtual void | calc_individual_fitness_from_traits (clone_t &tempgt) |
| Calculate fitness from traits of the chosen clone. More... | |
| virtual void | calc_individual_fitness_from_traits (int clonenum) |
| void | add_clone_to_genealogy (int locus, int dest, int parent, int left, int right, int cs, int n) |
Protected Attributes | |
| gsl_rng * | evo_generator |
| gsl_rng * | label_generator |
| int | seed |
| vector< int > | random_sample |
| int | number_of_loci |
| int | population_size |
| int | number_of_traits |
| int | generation |
| int | number_of_clones |
| double | mutation_rate |
| double | participation_ratio |
| int | last_clone |
| bool | allele_frequencies_up_to_date |
| double * | allele_frequencies |
| double * | gamete_allele_frequencies |
| double * | chi1 |
| double ** | chi2 |
| bool | all_polymorphic |
| vector< int > | ancestral_state |
| vector< poly_t > | polymorphism |
| vector< poly_t > | fixed_mutations |
| vector< int > | number_of_mutations |
| double | outcrossing_rate_effective |
| int * | genome |
| int * | crossovers |
| vector< int > | sex_gametes |
| double | fitness_max |
| stat_t | fitness_stat |
| stat_t * | trait_stat |
| double ** | trait_covariance |
| double * | trait_weights |
| bool | track_genealogy |
Population class for high-dimensional simulations.
This class is the main object storing the state of and enabling the manipulation of populations with long genomes ( \(L\)) larger than 20.
Both asexual and sexual populations can be simulated. Since asexual populations under selection are often structured as a small list of large clones, the class stores the clones and their sizes instead of the individuals.
Class methods give access to a variety of information on the population, including:
| haploid_highd::haploid_highd | ( | int | L_in = 0, |
| int | rng_seed = 0, |
||
| int | n_o_traits = 1, |
||
| bool | all_polymorphic_in = false |
||
| ) |
Default constructor.
The population objects are initialized, but neither the random generator is initialized nor the memory for traits, gametes etc. is allocated. Please call set_up() before using the class.
Note: The sequence is assumed to be linear (not circular). You can change this by hand if you wish so.

|
virtual |
Destructor.
Memory is released here.
|
protected |
|
inline |
| void haploid_highd::add_genotype | ( | boost::dynamic_bitset<> | genotype, |
| int | n = 1 |
||
| ) |
Add the genotype specified by a bitset to the current population in in n copies.
| genotype | genotype being added |
| n | number of copies of that genotype |
Note: this function also calculates the traits and fitness of the new individual.
|
protected |
Pair and mate sexual gametes.
Using the previously produced list of sex_gametes, pair them at random and mate
|
inline |

| int haploid_highd::bottleneck | ( | int | size_of_bottleneck | ) |
Cause a bottleneck in the population size.
| size_of_bottleneck | number of individuals to leave alive (approximate) |
The bottleneck is performed as follows. Each clone is traded in for a smaller one with size given by a Poisson random number around the new population size times the clone frequency. This function should therefore almost conserve genotype frequencies, except for rare genotypes that are lost.

|
protected |
calculate and store allele frequencies
Note: the allele frequencies are available in the allele_frequencies attribute.

|
protected |
Calculate and store fitness population statistics.
The fitness statistics are stored in fitness_stat.
Note: this function assumes that fitness is up to date. If you are not sure, call update_traits() and update_fitness() first.
|
protected |
Calculate fitness of a particular clone.
| tempgt | clone whose fitness is being calculated |
Note: this function also updates the traits information for the same clone, because the phenotype is needed to calculate fitness. If you have already calculated the traits, you can rely calc_individual_fitness_from_traits.
|
inlineprotected |
|
protectedvirtual |
Calculate fitness from traits of the chosen clone.
| tempgt | clone whose fitness is to be calculated |
This function is linear in the traits with weights equal to trait_weights. By default, only the first weight is different from zero.
|
inlineprotectedvirtual |


|
protected |
Calculate traits of the chosen clone.
| tempgt | clone whose traits are to be calculated |
|
inlineprotected |
| void haploid_highd::calc_stat | ( | ) |
Calculate trait and fitness statistics and allele frequences.
Four things are done in a row:
Note: This function is quite expensive. Please use its subblocks whenever possible, and rely on this only when you want to make sure, at the expense of performance, that everything is up to date.
|
protected |
Calculate and store trait population statistics and covariances.
The traits statistics are stored in traits_stat.
Note: this function assumes that traits are up to date. If you are not sure, call update_traits() first.
|
inlineprotected |
|
inline |
|
inline |
|
inline |
|
protected |
Choose a number of crossover points and produce a crossover pattern.
A typical crossover pattern would be 0000111100011111101101.

|
inline |
| int haploid_highd::distance_Hamming | ( | boost::dynamic_bitset<> | gt1, |
| boost::dynamic_bitset<> | gt2, | ||
| vector< unsigned int * > * | chunks = NULL, |
||
| unsigned int | every = 1 |
||
| ) |
Calculate Hamming distance between two sequences.
| gt1 | first sequence |
| gt2 | second sequence |
| chunks | (pointer to) vector of ranges (C pairs), e.g. ((0,10), (25, 28), (68, 70)) |
| every | check only every X sites, starting from the first of each chunk |
Note: you cannot use every without chunks.
Note: every chunk is a C array of length 2, i.e. a pointer to lower boundary of the interval. Thus we have the following situation:
chunks is of type vector <unsigned int *> **chunks is of type vector <unsigned int *>(*chunks)[i] is of type *unsigned int(*chunks)[i][0] is of type unsigned int, and indicates the initial site for this chunk(*chunks)[i][1] is of type unsigned int, and indicates the (final site +1) for this chunkAs a consequence, for each chunk, chunk[1] - chunk[0] is the length of the chunk.
When you prepare the vector of chunks for this function, please use C++ methods (new) that take care of memory management, or be very careful about memory leaks.
Note: this function is overloaded with simpler arguments (e.g. if you want to use clone indices).
| int haploid_highd::evolve | ( | int | gen = 1 | ) |
Evolve for some generations under the specified conditions.
The order of steps performed is
| gen | number of generations. |
Note: if an error in encountered, evolution is stopped after the function that created the problem. Typical errors include extinction or the opposite, offspring explosion.
| unsigned int haploid_highd::flip_single_locus | ( | int | locus | ) |
Flip a spin at a specific locus in random individual.
| locus | position of the locus to flip |
Note: this function calls flip_single_locus(unsigned int clonenum, int locus).


|
protected |
Flip a spin (locus) in individual.
| clonenum | the individual whose locus is being flipped |
| locus | position of the locus to flip |
This function creates a new clone and adds it to the population, and assigns it a fitness.
Note: This might produce duplicate clones since the mutant clone produced might already exist. Duplicates can be merged by the member unique_clones()
|
inline |
|
inline |
|
inline |
|
inline |
|
inline |
|
inline |
| int haploid_highd::get_divergence_histogram | ( | gsl_histogram ** | hist, |
| unsigned int | bins = 10, |
||
| vector< unsigned int * > * | chunks = NULL, |
||
| unsigned int | every = 1, |
||
| unsigned int | n_sample = 1000 |
||
| ) |
Get histogram of divergence from the [00...0] bitset.
| hist | pointer to the gsl_histogram to fill |
| bins | number of bins in the histogram |
Note: an antialiasing algorithm is used to enforce integer bin widths. Note: because of the antialiasing requirement, the last bin might be underrepresented. Note: the number of used bins in the output might be smaller than requested, depending on sample size and antialiasing.
| chunks | (pointer to) vector of ranges (C pairs), e.g. ((0,10), (25, 28), (68, 70)) |
| every | check only every X sites, starting from the first of each chunk |
Note: you cannot use every without chunks.
Note: every chunk is a C array of length 2, i.e. a pointer to lower boundary of the interval. Thus we have the following situation:
chunks is of type vector <unsigned int *> **chunks is of type vector <unsigned int *>(*chunks)[i] is of type *unsigned int(*chunks)[i][0] is of type unsigned int, and indicates the initial site for this chunk(*chunks)[i][1] is of type unsigned int, and indicates the (final site +1) for this chunkAs a consequence, for each chunk, chunk[1] - chunk[0] is the length of the chunk.
| n_sample | size of the random sample to use (the whole population is often too large) |
| stat_t haploid_highd::get_divergence_statistics | ( | unsigned int | n_sample = 1000 | ) |
Calculate mean and variance of the divergence from the [00...0] bitset.
| n_sample | size of the statistical sample to use (the whole pop is often too large) |

| int haploid_highd::get_diversity_histogram | ( | gsl_histogram ** | hist, |
| unsigned int | bins = 10, |
||
| vector< unsigned int * > * | chunks = NULL, |
||
| unsigned int | every = 1, |
||
| unsigned int | n_sample = 1000 |
||
| ) |
Get histogram of diversity in the population (mutual Hamming distance)
| hist | pointer to the gsl_histogram to fill |
| bins | number of bins in the histogram |
Note: an antialiasing algorithm is used to enforce integer bin widths. Note: because of the antialiasing requirement, the last bin might be underrepresented. Note: the number of used bins in the output might be smaller than requested, depending on sample size and antialiasing.
| chunks | (pointer to) vector of ranges (C pairs), e.g. ((0,10), (25, 28), (68, 70)) |
| every | check only every X sites, starting from the first of each chunk |
Note: you cannot use every without chunks.
Note: every chunk is a C array of length 2, i.e. a pointer to lower boundary of the interval. Thus we have the following situation:
chunks is of type vector <unsigned int *> **chunks is of type vector <unsigned int *>(*chunks)[i] is of type *unsigned int(*chunks)[i][0] is of type unsigned int, and indicates the initial site for this chunk(*chunks)[i][1] is of type unsigned int, and indicates the (final site +1) for this chunkAs a consequence, for each chunk, chunk[1] - chunk[0] is the length of the chunk.
| n_sample | size of the random sample to use (the whole population is often too large) |
| stat_t haploid_highd::get_diversity_statistics | ( | unsigned int | n_sample = 1000 | ) |
Calculate diversity in the current population (Hamming distance between pairs of sequences)
| n_sample | size of the statistical sample to use (the whole pop is often too large) |
|
inline |
| int haploid_highd::get_fitness_histogram | ( | gsl_histogram ** | hist, |
| unsigned int | bins = 10, |
||
| unsigned int | n_sample = 1000 |
||
| ) |
Calculate histogram of fitness from traits.
| hist | pointer to the gsl_histogram to fill |
| bins | number of bins in the histogram |
Note: the output histogram might have less bins than requested if the sample size is too small.
| n_sample | size of the random sample to use (the whole population is often too large) |
There is a small problem here, namely that sometimes the fitness distribution has a horrible tail which messes up the calculation of the bin width. Thus we first calculate the fitness average and variance, and then set the bin width so that the deleterious tail is within 2 sigma or so.
Note: this function allocates memory for the histogram only if there is no binning error. If you get HP_NOBINSERR, no memory is allocated. The user is expected to release the memory manually.
|
inline |
|
inline |
|
inline |

|
inline |
|
inline |
|
protected |
Get the population exp-average of fitness, used for keeping the population size fixed.
Mathematically, this is \( \log\left( \left< e^(F-F_{max}) \right> \right) \). The baseline is \( F_{max} \) in order to avoid exponentiating large numbers.

|
inline |
|
inline |
|
inline |
| vector< int > haploid_highd::get_nonempty_clones | ( | ) |
Obtain a list of the good clones.
|
inline |
|
inline |
|
inline |
|
inline |
| vector< double > haploid_highd::get_pair_frequencies | ( | vector< vector< int > > * | loci | ) |
Get the joint frequency of two alleles, for a vector of allele pairs.
| loci | pointer to a vector of allele pairs. Each element of loci must be a vector of length 2. |
| double haploid_highd::get_pair_frequency | ( | int | locus1, |
| int | locus2 | ||
| ) |
Get the joint frequency of two alleles.
| locus1 | position of the first allele. |
| locus2 | position of the second allele. |
|
inline |
|
inline |
|
inline |
|
protected |
Get a random seed from /dev/urandom.
|
inline |
|
inline |
|
protected |
Calculate trait difference between two clones.
| tempgt1 | first clone |
| tempgt2 | second clone |
| diffpos | positions at which the genotypes differ |
| traitnum | number of the trait to calculate |
The operation is, for each trait, tempgt1 - tempgt2.
|
inline |
|
inline |
|
inline |
|
inline |
|
inline |
|
protected |
Mutate random clones at random loci (all loci)
Note: for efficiency reasons, if the mutation rate is zero, an if cycle skips the body altogether. The user can therefore call this function safely even if in non-mutating populations, without loss of performance.
Function is now rewritten to allow for multiple mutants. TODO: Poisson conditional on at least one FIXME!: all_polymorphic assumes that all additive effects are set, i.e. that locus equals the index of the coefficient in the vector of additive effects

|
inline |
|
protected |
|
protected |
Calculate the cumulative partition of sequences into clones.
| partition_cum | the vector to be filled |
Example: if there are three clones of sizes (100, 22, 3) this function will fill the vector (100, 122, 125). The last element is of course the population size See also get_population_size.
Note: the vector is taken in input by reference for performance reasons, since it can get huge.
| int haploid_highd::print_allele_frequencies | ( | ostream & | out | ) |
Print all allele frequencies into a stream provided.
| out | stream to put the allele frequencies (usually a file or stdout) |
|
protected |
Produce and store a random sample of the population for stochastic processes.
| size | size of the sample |
Produce a random sample of genotypes (labeled by their clone of origin) which is to be used for mutations and sampling. The vector random_sample exists for efficiency reasons, for it is faster to call the random number generator once than many times.
Note: if you want to get random clones, please use random_clone().
|
protected |
: allocates memory for a sufficient number of clones
: number of empty clones required


| int haploid_highd::random_clone | ( | ) |
Get a random clone from the population.
The probability density function from which the individual is chosen is flat over the population (larger clones are proportionally more likely to be returned here).
Note: for efficiency reasons, the class keeps a storage of random clone indices. If you need much more than, say, 1000 random clones at a time, please call produce_random_sample with the required size in advance.
| int haploid_highd::random_clones | ( | unsigned int | n_o_individuals, |
| vector< int > * | sample | ||
| ) |
Sample random individuals from the population.
| n_o_individuals | number of individuals to sample |
| sample | pointer to vector where to put the result |
The results are not returned as a vector for performance reasons, as one might want to get a lot of random clones. *sample may be not empty (but must be allocated). In any case, clone numbers of the sampled individuals are appended to *sample. Hence, you can use this function iteratively (although there might not be a good reason to do so).


| int haploid_highd::read_ms_sample | ( | istream & | gts, |
| int | skip_locus, | ||
| int | multiplicity | ||
| ) |
Read the output of Hudson's ms and use it to initialize the genotype distribution.
| gts | genotypes output of ms |
| skip_locus | position of the locus to be skipped |
| multiplicity | number of times each genotype is added |
ms loci are fed into the genotype with a locus that is skipped. Each ms genotype is added multiple times.

| int haploid_highd::read_ms_sample_sparse | ( | istream & | gts, |
| int | skip_locus, | ||
| int | multiplicity, | ||
| int | distance | ||
| ) |
Read the output of Hudson's ms and use it to initialize the genotype distribution.
| gts | genotypes output of ms |
| skip_locus | positionof the locus to be skipped |
| multiplicity | number of times each genotype must be added |
| distance | distance between two polymorphic sites |
ms loci are fed into the genotype at distance "distance", i.e. there are distance-1 monomorphic loci. One locus is skipped. Each ms genotype is added multiple times.
|
protected |
Produce a random reassortement pattern.

|
protected |
Recombine two genotypes parent1 and parent2 to produce two new genotypes.
| parent1 | first parent |
| parent2 | second parent |
The new genotypes are stored in new_pop at positions ng and ng+1.

|
protected |
|
protected |
Get the log of the exp-average fitness plus relaxation term.
|
protected |
Generate offspring according to fitness (selection) and segregate some for sexual mating.
Random Poisson offspring numbers are drawn from all parents, proportionally to their fitness. A fraction r of all offspring are designated for sexual reproduction, while the remainder (1-r) reproduces by exact duplication (mutations are introduced later). The population size relaxes to a carrying capacity, i.e. selection is soft but the population size is not exactly fixed.

| int haploid_highd::set_allele_frequencies | ( | double * | freq, |
| unsigned long | N_in | ||
| ) |
Initialize population in linkage equilibrium.
| freq | target allele frequencies |
| N_in | number of individuals to be created |
Note: the carrying capacity is set equal to N_in if it is still unset.
|
inline |
| int haploid_highd::set_genotypes | ( | vector< genotype_value_pair_t > | gt | ) |
Initialize the population with genotype counts.
| gt | vector of genotype_value_pair with genotypes and sizes |
Note: the population size is set as the total sum of counts of all genotypes. The carrying capacity is also set to the same number if it is still unset.
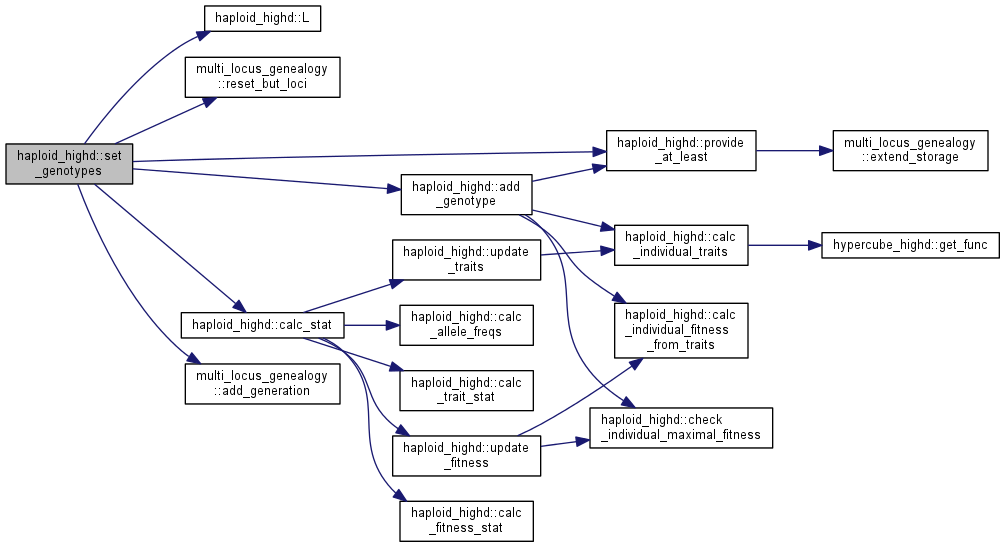
|
inline |
|
inline |
|
inline |
|
inline |
| int haploid_highd::set_wildtype | ( | unsigned long | N_in | ) |
Initialize a wildtype population (00...0)
| N_in | number of individuals |
Note: the carrying capacity is set equal to N_in if it is still unset.
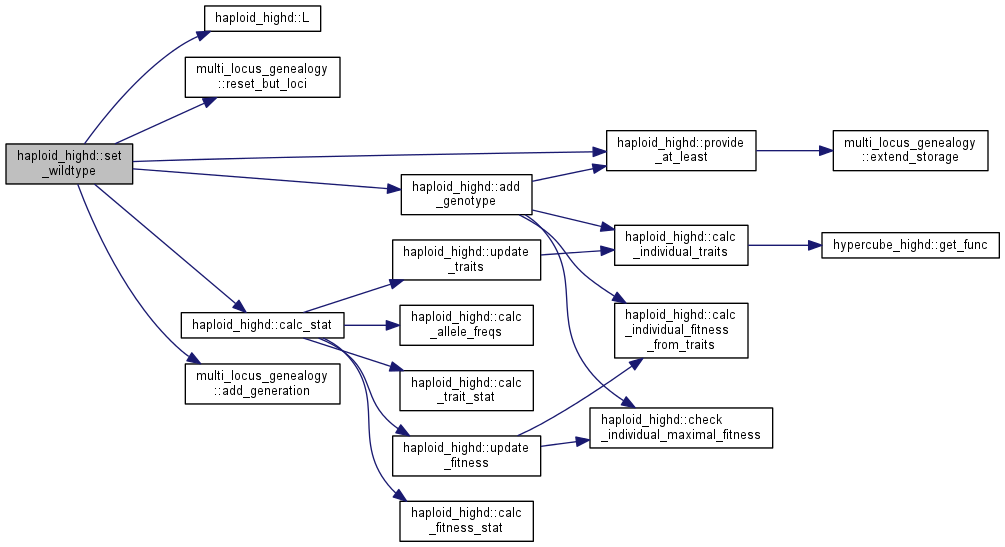
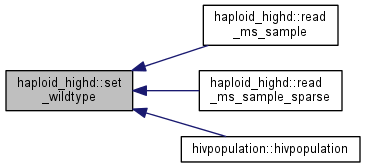
|
protected |
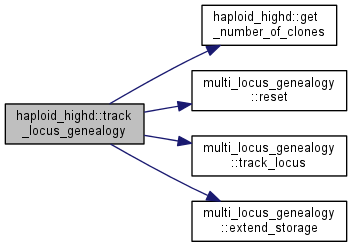
| int haploid_highd::track_locus_genealogy | ( | vector< int > | loci | ) |
Designates as set of loci to have their genealogy tracked.
locus to be tracked
| void haploid_highd::unique_clones | ( | ) |
Remove duplicate clones.
The library does not usually check whether two clones with the same genotype are present, but this can happen in case of recurrent mutation. This function merges duplicates.
Note: this is only needed for studying the clone structure. Evolution itself does not need to make sure that clones are unique.
| void haploid_highd::update_fitness | ( | ) |
For each clone, update fitness assuming traits are already up to date.


| void haploid_highd::update_traits | ( | ) |
For each clone, recalculate its traits.

|
protected |
|
protected |
|
protected |
|
protected |
| int haploid_highd::carrying_capacity |
|
protected |
|
protected |
| bool haploid_highd::circular |
| double haploid_highd::crossover_rate |
|
protected |
|
protected |
|
protected |
|
protected |
|
protected |
|
protected |
| multi_locus_genealogy haploid_highd::genealogy |
|
protected |
|
protected |
| double haploid_highd::growth_rate |
|
protected |
|
protected |
|
protected |
|
protected |
|
protected |
|
protected |
|
protected |
| double haploid_highd::outcrossing_rate |
|
protected |
|
protected |
|
protected |
| vector<clone_t> haploid_highd::population |
|
protected |
|
protected |
| int haploid_highd::recombination_model |
|
protected |
|
protected |
|
protected |
| hypercube_highd* haploid_highd::trait |
|
protected |
|
protected |
|
protected |
 1.8.3.1
1.8.3.1 
