
Overview of FFPopSim
FFPopSim consists of two parts enabling genotype-based or individual-based simulations. The class "haploid_lowd" can be used for simulations of a few sites (up to 20) and models the distribution of the population among all possible genotypes. "haploid_highd" allows to simulate longer genomes by modeling by tracking only the genomes present in the population.Both classes include a rich variety of utility functions, and can be extended by C++/Python subclassing seamlessly. Documentation and examples of C++ routines, Python scripts, and subclassing are included.
Genotype-based simulations: haploid_lowd
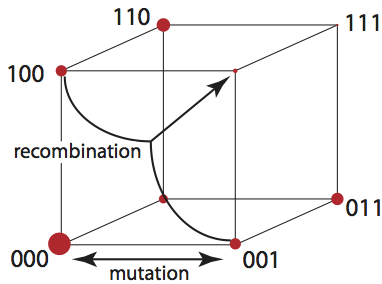 The binary hypercube is used to represent genotypes in "haploid_lowd".
Mutation is equivalent to a flow along edges of the cube, recombination takes two
genomes (vertices of the cube) and produces new genomes be mixing the parental genomes.
The binary hypercube is used to represent genotypes in "haploid_lowd".
Mutation is equivalent to a flow along edges of the cube, recombination takes two
genomes (vertices of the cube) and produces new genomes be mixing the parental genomes.
"haploid_lowd" models the whole genotype distribution efficiently, offering the following features:
- Tracks the frequencies of all possible genotypes
- Arbitrarily large populations
- Arbitrary fitness functions and genetic maps (including time-dependent)
- Asexual and sexual populations
- Low run-time complexity (compared to the naive O(8^L)):
- O(L 2^L) with single crossover recombination -> up to 20 loci.
- O(3^L) with general recombination -> up to 15 loci.
- Site-specific recombination rates, site- and direction-specific mutation rates
- Deterministic and stochastic simulations
Individual-based simulations: haploid_highd
 Genomes are represented in "haploid_highd" as bitstrings, ensuring an
efficient memory usage. Mutation is a bit flip, and recombination creates
a new bitstring from two existing ones.
Genomes are represented in "haploid_highd" as bitstrings, ensuring an
efficient memory usage. Mutation is a bit flip, and recombination creates
a new bitstring from two existing ones."haploid_highd" is a more traditional, individual-based simulation interface for populations with a larger number of sites. It uses clones instead of individuals to increase efficiency in case of low mutation and recombination rates; it is not, however, restricted to these conditions, and is currently used to simulate the highly variable Human Immunodeficieny Virus (HIV). It offers the following features:
- Long genomes (several millions of loci, depending on memory availability)
- Large populations (e.g. 100,000 individuals with 1,000,000 loci each)
- Tracks several phenotypic traits that codetermine fitness
- Arbitrary genotype-phenotype maps (including time-dependent)
- Asexual and sexual populations